注:本文未经金阁顿授权禁止转载,否则将视为侵权,我们将采取法律措施维护权益。
新加坡根据《健康产品法》(Health Products Act,简称HPA)及其配套的《健康产品(医疗器械)条例》(Health Products (Medical Devices) Regulations)对医疗器械进行监管。新加坡卫生科学局(Health Sciences Authority,HSA)要求企业在制造、进口或供应医疗器械之前必须获得经销商牌照。除A类低风险医疗器械(可豁免产品注册)或经HSA批准的特殊情况外,所有医疗器械均需在HSA完成注册,方可在新加坡供应。
一、新加坡医疗器械产品的风险分类
每项器械注册均通过特定评估路径进行。器械的评估路径取决于以下因素:
● 器械的风险分类;
● 获得海外参考监管机构事先批准的数量;
● 器械的安全销售历史时长。
器械的评估路径将决定注册所需的处理时限、费用及文件要求。

1.通用医疗器械
(1)通用医疗器械风险分类体系
通用医疗器械分为四个风险等级,产品负责人必须根据每项医疗器械的预期用途来应用风险分类规则:
● A类风险:低风险,例如轮椅或压舌板;
● B类风险:低至中度风险,例如皮下注射针头或吸引设备;
● C类风险:中度至高风险,例如呼吸机或骨固定板;
● D类风险:高风险,例如心脏瓣膜或植入式除颤器。
(2)影响通用医疗器械风险分类的因素
影响通用医疗器械风险分类的因素有很多,其中包括:
● 医疗器械与人体接触的持续时间;
● 侵入程度;
● 医疗器械是否向患者输送药品或能量;
● 是否预期对患者产生生物学效应;
● 局部效应与全身效应(例如,非可吸收缝线与可吸收缝线)。
根据产品负责人的预期用途,若有两项或多项风险分类规则适用于该医疗器械,则应将该医疗器械归入最高的风险等级。
如果一件医疗器械预期与另一件医疗器械组合使用,则应分别对每件医疗器械应用风险分类规则。无论它们是否来自同一产品负责人(例如,生理监护仪与独立的记录仪,或通用注射器与注射泵),均应如此处理。

(3)通用医疗器械组合
对于单独已符合所有法规要求的医疗器械组合,其风险分类取决于产品负责人包装和销售的目的。例如:
● 如果组合后形成的产品旨在实现不同于其各组成部分医疗器械的用途,则该组合应被视为一个新的医疗器械,并应相应地进行分类;
● 如果组合仅是为了方便用户,并未改变其各组成部分医疗器械的预期用途(例如,提供执行特定外科手术所需所有医疗器械的定制套件),则该组合所分配的风险等级应与其中包含的最高风险等级的医疗器械相同,此分类用于编制符合性声明。
符合性声明


(4)与通用医疗器械配合使用的附件
预期专门与医疗器械配合使用以帮助其发挥功能的附件,应遵循适用于医疗器械本身的监管要求(例如,安全与性能基本原则、上市后监督等)。
(5)与通用医疗器械配合使用的软件
大多数软件内置于医疗器械本身(例如,操作心电图机的嵌入式软件)。
某些软件应用程序并未内置于医疗器械本身(例如,在独立于心电图机的计算机上分析心电图信号的软件应用程序)。这些被视为独立软件,属于医疗器械定义的范畴,并应如下分类:
● 若其驱动或影响单独医疗器械的使用,则应根据该组合的预期用途进行分类;
● 若其独立于任何其他医疗器械,则应根据规则自行分类;
● 独立软件被视为有源医疗器械。(注:有源医疗器械是指任何其运行依赖于电能或除人体直接产生或重力之外的任何能源,并通过转换此种能量而发挥作用的医疗器械。预期在有源医疗器械与患者之间传输能量、物质或其他元素,且不发生任何显著变化的医疗器械,不被视为有源医疗器械。)
有关通用医疗器械的风险分类规则,请参阅医疗器械指导文件中的GN-13:通用医疗器械风险分类指导,网址:
https://www.hsa.gov.sg/medical-devices/guidance-documents#toggle=togglepanel-product-registration。
2.体外诊断医疗器械
(1)体外诊断医疗器械分类体系
体外诊断医疗器械也分为四个风险等级,产品负责人必须根据每项医疗器械的预期用途来应用风险分类规则:
● A类风险:个人风险低且公共卫生风险低 ,例如标本容器;
● B类风险:中个人风险中等或公共卫生风险低,或二者兼具,例如维生素B12检测、妊娠自测试剂、抗核抗体检测、尿试纸条;
● C类风险:个人风险高或公共卫生风险中等,或二者兼具,例如血糖自测试剂、HLA分型、前列腺特异性抗原筛查、风疹病毒IgM检测;
● D类风险:个人风险高且公共卫生风险高,例如HIV献血者筛查试剂、HIV诊断试剂盒。

(2)影响体外诊断医疗器械风险分类的因素
影响体外诊断医疗器械的风险分类的因素有很多,其中包括:
● 确定产品在其预期用途和使用指征上是否符合体外诊断医疗器械的定义;
● 考虑正确的分类规则,如果体外诊断医疗器械根据产品负责人的规定具有多种预期用途,导致其可能归属于多个类别,则应将其归入较高的类别;
● 若有两项或多项风险分类规则适用于该体外诊断医疗器械,则应将其归入最高的风险等级;
● 将产品归入特定风险等级的理由应予以记录。
其他需要考虑的因素包括:
● 预期与体外诊断试剂一起使用的校准品,应按照与该试剂相同的风险等级进行处理;
● 具有为单一特定分析物或多个分析物赋值的定值质控品,应归入与相应体外诊断试剂相同的风险等级。
(3)与体外诊断医疗器械配合使用的软件
● 大多数软件内置于体外诊断医疗器械本身,例如,操作分析仪的嵌入式软件。对于此类软件,如果其控制或影响单独体外诊断医疗器械的预期输出,则其风险等级与体外诊断医疗器械本身相同;
● 有些软件并未内置于医疗器械本身,例如,根据分析仪结果提供分析的软件。此类软件被视为独立软件。当它不内置于体外诊断医疗器械时,应使用分类规则自行进行分类。
有关风险分类规则,请参阅医疗器械指导文件中的GN-14:体外诊断医疗器械风险分类指导,网址:
https://www.hsa.gov.sg/medical-devices/guidance-documents#toggle=togglepanel-product-registration。

二、新加坡医疗器械注册要求概览
1.A类器械注册
A类医疗器械可豁免产品注册。但是,对于您制造或进口的A类医疗器械,您需要在SHARE系统上提交产品通知(product notification)申请。
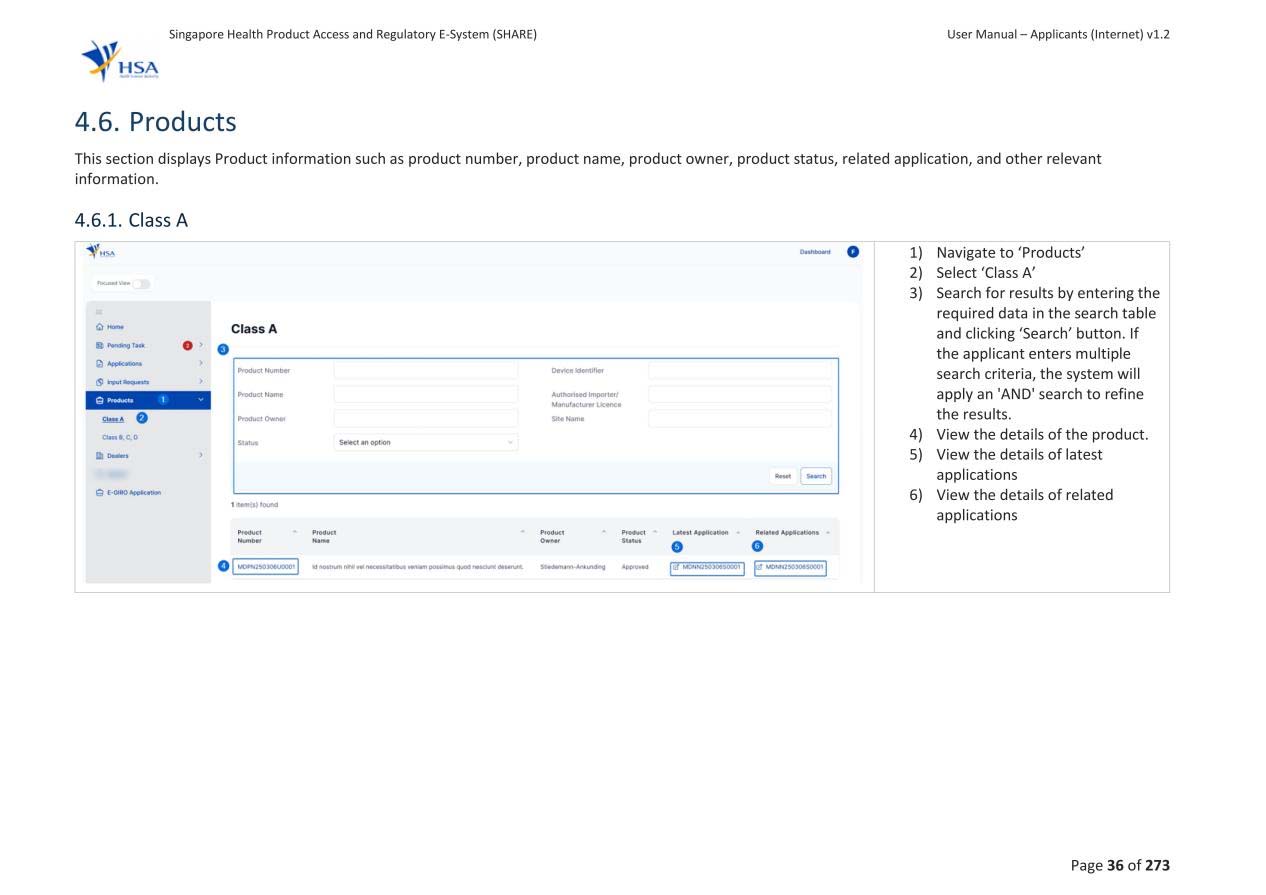
2.B类、C类及D类器械完整注册路径概述
完整注册(Full)
未获得任何海外参考监管机构事先批准的医疗器械,必须接受完整注册。
完整注册(优先审评计划)(Full (Priority Review Scheme))
优先审评计划为医疗器械提供更快的注册和市场准入通道,该计划仅适用于通过完整注册路径提交的B类、C类和D类器械,且不适用于含有次要注册药用物质的D类器械。与标准完整注册路径相比,优先审评计划的总处理时间缩短35%。该计划包含两条路径:
路径1
医疗器械需同时满足以下两项标准:
● 属于以下五个重点医疗领域之一:
○ 癌症;
○ 糖尿病;
○ 眼科疾病;
○ 心血管疾病;
○ 传染病;
● 其设计和验证旨在满足未满足的临床需求。这意味着该器械:
○ 可用于诊断或治疗一种尚无其他现有疗法的疾病;或
○ 是一项相较于现有技术具有显著优势的突破性技术。
路径2
医疗器械不符合上述两条标准。

3.海外参考监管机构
符合简化、加速及立即注册路径要求的批准机构如下所示,这些批准必须针对拟在新加坡市场销售的、具有相同标签用途的医疗器械:
澳大利亚-Therapeutic Goods Administration(TGA)
器械注册牌照。
欧洲-欧盟公告机构(EU Notified Bodies,EU NB)【注:欧盟医疗器械指令(MDD/AIMDD/IVDD)已废止,现行为医疗器械法规(MDR)和体外诊断医疗器械法规(IVDR)。HSA接受依据已废止指令签发的有效证书作为历史批准证明,但企业应关注欧盟现行法规。】
对于B类器械-依据以下签发的EC证书:
● 指令93/42/EEC附录II第3节或附录V(针对IIa类器械);
● 医疗器械法规附录IX第一章和第三章,或MDR附录XI PART A(针对IIa类器械);
● 指令98/79/EC附录IV或附录V结合附录VII(针对B类列表和自我检测用体外诊断医疗器械);
● 体外诊断医疗器械法规附录IX第一章和第三章(针对B类IVD)。
对于C类和D类器械-依据以下签发的EC证书:
● 指令93/42/EEC附录II第3节或附录III结合附录V(针对IIb类)
● 医疗器械法规附录IX第一章和第三章(包括对植入式器械技术文件的评估),或MDR附录X结合附录XI PART A(针对IIb类)
● 指令93/42/EEC附录II第3节和第4节(针对III类)
● 医疗器械法规附录IX第一章和第三章(包括对III类器械技术文件的评估)
● 指令90/385/EEC附录II第3节和第4节(针对有源植入式医疗器械)(注:指令90/385/EEC已并入MDR,有源植入式医疗器械在MDR下属于III类)
● 指令98/79/EC附录IV(包括第4和第6节)(针对A类列表IVD)
● 体外诊断医疗器械法规附录IX第一章和第三章(包括对D类IVD技术文件的评估)
● 指令98/79/EC附录IV或附录V结合附录VII(针对B类列表和自我检测用体外诊断医疗器械)
● 体外诊断医疗器械法规附录IX第一章和第三章(包括对伴随诊断、自我检测和近患者检测器械技术文件的评估),或IVDR附录X结合附录XI(第5节除外)(针对C类IVD)
加拿大-Health Canada(HC)
器械注册牌照。
日本-厚生劳动省(Ministry of Health, Labour and Welfare,MHLW)
● 日本注册认证机构颁发的上市前认证;
● 厚生劳动省颁发的上市前批准。
美国-Food and Drug Administration (US FDA)
● 510(K)许可;
● De Novo分类;
● 上市前批准。
注意:在上述参考监管机构被分类并批准/许可为I类或II类豁免的B类、C类和D类医疗器械,基于这些相应的批准,不符合简化、加速或立即注册路径的资格。

三、新加坡医疗器械注册指南
1.B类医疗器械注册指南
(1)B类医疗器械完整注册
资格要求
若您的医疗器械此前未获得任何独立的海外参考监管机构批准,则必须通过完整注册路径进行注册。
优先审评计划
优先审评计划为医疗器械公司提供了加快注册和市场准入的选项,您可以在SHARE系统中选择加入此计划。
提交要求
所有文件均需以英文提交,此提交要求同样适用于选择优先审评计划的申请人。
东盟统一提交资料模板(CSDT)文件清单
CSDT为向东盟成员国医疗器械监管机构提交医疗器械信息提供了一个通用模板。您需要提交以下CSDT文件作为产品注册要求的一部分:
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件的详细信息:
○ 临床前研究的完整报告(例如,物理测试数据、生物相容性研究、动物研究以及软件验证与确认研究);
○ 计量要求;
○ 灭菌验证;
○ 保质期研究及预期使用寿命;
● 临床评估报告,包括所引用研究的出版物和完整报告;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
若器械标注的用途超出其常规用途,可能会被要求提供进一步的临床数据支持。
补充文件
除CSDT文件外,您还需提交以下文件:
● 授权书;
● 医疗器械配置清单。

(2)B类医疗器械简化注册
资格要求
如果您的B类医疗器械目前已获得至少一个独立的海外参考监管机构的批准,并且该批准所针对的器械标签用途与拟在新加坡市场销售的用途相同,则您有资格通过简化注册路径进行注册。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件总结:
○ 临床前研究总结(包括灭菌验证和货架寿命研究,如适用);
● 临床评估报告(如适用);
● 医疗器械标签;
● 风险分析;
● 制造商信息:
○ 制造场所和灭菌场所的名称与地址;
○ 至少符合下列质量管理体系之一的证明:
⊕ ISO 13485;
⊕ MDSAP;
⊕ 符合美国FDA质量体系法规;
⊕ 日本厚生劳动省第169号法令。
若器械标注的用途超出其常规用途,可能会被要求提供进一步的临床数据支持。
补充文件
除CSDT文件外,您还需提交以下文件:
● 授权书;
● 医疗器械配置清单;
● 获得独立的海外参考监管机构批准的证明(例如,批准信函、证书)。

(3)B类医疗器械立即注册
资格要求
如果您的B类医疗器械符合以下条件,则有资格通过立即注册路径进行注册:
条件1
● 获得至少一个独立的海外参考监管机构的批准;
● 该批准所针对的器械标签用途必须与拟在新加坡市场销售的用途相同;
● 已在至少一个独立的海外参考监管机构管辖区域内上市销售至少三年;
● 该医疗器械未曾因质量、性能或安全性问题被任何独立的海外参考监管机构拒绝或撤销批准;
● 在过去3年内(或自该医疗器械在全球上市以来),全球范围内无与该医疗器械使用相关的安全性问题。具体定义为:
○ 无死亡报告;
○ 无导致任何人健康状况严重恶化的报告;
○ 在提交申请时,无未解决的现场安全纠正措施(包括召回)。
条件2
● 获得至少两个独立的海外参考监管机构的批准;
● 这些批准所针对的器械标签用途必须与拟在新加坡市场销售的用途相同;
● 该医疗器械未曾因质量、性能或安全性问题被任何独立的海外参考监管机构拒绝或撤销批准;
● 在过去三年内,全球范围内无与该医疗器械使用相关的安全性问题,包括:
○ 无死亡报告;
○ 无导致任何人健康状况严重恶化的报告;
○ 在提交申请时,无未解决的现场安全纠正措施(包括召回)。

独立医疗移动应用程序
● 该器械仅为独立的医疗移动应用程序;
● 获得至少一个独立的海外参考监管机构的批准;
● 该批准所针对的器械标签用途必须与拟在新加坡市场销售的用途相同;
● 该医疗器械未曾因质量、性能或安全性问题被任何独立的海外参考监管机构拒绝或撤销批准;
● 在过去三年内,全球范围内无与该医疗器械使用相关的安全性问题,包括:
○ 无死亡报告;
○ 无导致任何人健康状况严重恶化的报告;
○ 在提交申请时,无未解决的现场安全纠正措施(包括召回)。
注意:如果您的B类医疗器械同时获得欧盟和TGA的批准,将仅视为一个批准。
请确保您确实满足上述所有条件,因为通过立即注册路径提交申请成功后,您的医疗器械将被纳入新加坡医疗器械注册簿。若资格不符合要求,将导致您的器械注册无效,并且您需要重新通过其他注册路径申请注册(需支付相应费用)。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 医疗器械描述;
● 临床前研究总结:
○ 仅限无菌器械提供灭菌验证;
○ 仅限独立医疗移动应用程序提供软件验证与确认研究;
○ 提供支持联网医疗器械网络安全的证据;
● 医疗器械标签;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令。
若器械标注的用途超出其常规用途,可能会被要求提供进一步的临床数据支持。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得独立的海外参考监管机构批准的证明(例如,批准信函、证书);
● (仅适用于B类立即注册条件1)在相同独立的海外参考监管机构管辖区域内的市场营销历史证明:
○ 带日期的发票;
○ 销售证明;
○ 市场营销历史声明;
● 全球无安全性问题声明。

2.C类医疗器械注册指南
(1)C类医疗器械完整注册
资格要求
若您的医疗器械此前未获得任何独立的海外参考监管机构批准,则必须通过完整注册路径进行注册。
优先审评计划
优先审评计划为医疗器械公司提供了加快注册和市场准入的选项,您可以在SHARE系统中选择加入此计划。
提交要求
所有文件均需以英文提交,此提交要求同样适用于选择优先审评计划的申请人。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件的详细信息:
○ 临床前研究的完整报告(例如,物理测试数据、生物相容性研究、动物研究以及软件验证与确认研究);
○ 计量要求;
○ 灭菌验证(如适用);
○ 保质期研究及预期使用寿命;
● 临床评估报告,包括所引用研究的出版物和完整报告;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
○ 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单。

(2)C类医疗器械简化注册
资格要求
如果您的C类医疗器械目前已获得至少一个独立的海外参考监管机构的批准,并且该批准所针对的器械标签用途与拟在新加坡市场销售的用途相同,则您有资格通过简化注册路径进行注册。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件总结:
○ 临床前研究总结(包括灭菌验证和货架寿命研究,如适用);
● 临床评估报告;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得独立的海外参考监管机构批准的证明(例如,批准信函、证书)。
(3)C类医疗器械立即注册
资格要求
如果您的C类医疗器械符合以下条件,则有资格通过立即注册路径进行注册:
● 该器械仅为独立的医疗移动应用程序;
● 获得至少一个独立的海外参考监管机构的批准;
○ 该批准所针对的器械标签用途必须与拟在新加坡市场销售的用途相同;
● 该医疗器械未曾因质量、性能或安全性问题被任何独立的海外参考监管机构拒绝或撤销批准;
● 在过去三年内,全球范围内无与该医疗器械使用相关的安全性问题,包括:
○ 无死亡报告;
○ 无导致任何人健康状况严重恶化的报告;
○ 在提交申请时,无未解决的现场安全纠正措施(包括召回)。
重要提示:如果在提交过程中,经判定您的申请不符合所选注册路径的资格要求,您将需要撤回当前申请,并依据正确的注册路径重新提交。先前支付的费用不予退还。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 医疗器械描述;
● 临床前研究总结:
○ 仅限独立的医疗移动应用程序提供软件验证与确认研究;
○ 提供支持联网医疗器械网络安全的证据;
● 医疗器械标签;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令。
若器械标注的用途超出其常规用途,可能会被要求提供进一步的临床数据支持。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得独立的海外参考监管机构批准的证明(例如,批准信函、证书);
● 全球无安全性问题声明。

3.D类医疗器械注册指南
(1)D类医疗器械完整注册
资格要求
若您的医疗器械此前未获得任何独立的海外参考监管机构批准,则必须通过完整注册路径进行注册。
优先审评计划
优先审评计划为医疗器械公司提供了加快注册和市场准入的选项,您可以在SHARE系统中选择加入此计划。
提交要求
所有文件均需以英文提交。此提交要求同样适用于选择优先审评计划的申请人。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件的详细信息:
○ 临床前研究的完整报告(例如,物理测试数据、生物相容性研究、动物研究以及软件验证与确认研究);
○ 灭菌验证(如适用);
○ 保质期研究及预期使用寿命;
● 临床评估报告,包括所引用研究的出版物和完整报告;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单。

(2)D类医疗器械简化注册
资格要求
如果您的D类医疗器械目前已获得至少一个独立的海外参考监管机构的批准,并且该批准所针对的器械标签用途与拟在新加坡市场销售的用途相同,则您有资格通过简化注册路径进行注册。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件总结:
○ 临床前研究总结(包括灭菌验证和货架寿命研究,如适用);
● 临床评估报告总结;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得独立的海外参考监管机构批准的证明(例如,批准信函、证书)。

(3)D类医疗器械加急注册
资格要求
如果您的D类医疗器械目前已获得至少两个独立的海外参考监管机构的批准,并且这些批准所针对的器械标签用途与拟在新加坡市场销售的用途相同,则您有资格通过加急注册路径进行注册。
重要提示:来自欧盟和澳大利亚药品管理局的批准,仅在该器械已由相应监管机构独立审评并获得批准(而非基于《互认协定》进行注册)的情况下,方可被认定为独立的海外参考监管机构批准。
不符合EDR资格的器械
以下D类器械不具备通过加急注册路径注册的资格:
● 有源植入式器械(例如,心脏起搏器、神经刺激器);
● 与中枢循环系统直接接触的植入式器械;(注:主要的体内大血管包括但不限于以下血管:arteriae pulmonales(pulmonary artery)、aorta ascendens、(ascending aorta)arteriae coronariae(coronary artery)、arteria carotis communis(common carotid artery)、arteria carotis externa(external carotid artery)、arteria carotis interna(internal carotid artery)、arteriae cerebrates(cerebella arteries)、truncus brachicephalicits(brachiocephalic trunk)、venae cordis(cardiac veins)、venae pulmonales(pulmonary vein)、venae cava superior(superior vena cava)、venae cava inferior(inferior vena cava)、arcus aorta(aortic arch)、thoracica aorta(thoracic aorta)、abdominalis aorta(abdominal aorta)、ilica communis(common iliac arteries and veins)、aorta descendens to the bifurcatio aortae.(descending aorta to the bifurcation of aorta);
● 与中枢神经系统直接接触的植入式器械;(注:中枢神经系统是指大脑、脑膜及脊髓)
● 髋、膝和肩关节置换器械(例如,生物活性植入物);
● 含有次要注册药用物质的器械;
● 用于以下用途的体外诊断医疗器械:
○ HIV检测(筛查和诊断);
○ 血液/组织供体相容性测试。
上述器械仅能通过完整或简化注册路径进行注册。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件总结:
○ 临床前研究总结(包括灭菌验证和货架寿命研究,如适用);
● 临床评估报告总结;
● 医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明(例如):
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得两个独立的海外参考监管机构批准的证明(例如,批准信函、证书)。

(4)含有次要注册药用物质的D类医疗器械完整注册
资格要求
如果您的含有次要注册药用物质的D类医疗器械,此前未获得任何独立的海外参考监管机构的批准,并且其化学或生物成分也未经世界卫生组织认可的主管药品监管机构评估和批准,则必须通过完整注册路径进行注册。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件的详细信息:
○ 临床前研究的完整报告(例如,物理测试数据、生物相容性研究、动物研究以及软件验证与确认研究);
○ 灭菌验证(如适用);
○ 保质期研究及预期使用寿命;
● 临床评估报告,包括所引用研究的出版物和完整报告;
● 拟用医疗器械标签;
● 风险分析;
● 制造场所和灭菌场所的名称与地址;
● 至少符合下列质量管理体系之一的证明:
○ ISO 13485;
○ MDSAP;
○ 符合美国FDA质量体系法规;
○ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单。

(5)含有次要注册药用物质的D类医疗器械简化注册
资格要求
如果您的含有次要注册药用物质的D类医疗器械符合以下条件,则有资格通过简化注册路径进行注册:
● 目前已获得至少一个独立的海外参考监管机构的批准;并且
● 其化学或生物成分已经过世界卫生组织认可的主管药品监管机构评估和批准;
● 该批准所针对的器械标签用途必须与拟在新加坡市场销售的用途相同。
提交要求
所有文件均需以英文提交。
东盟统一提交资料模板(CSDT)文件清单
● 执行摘要;
● 基本要求检查表;
● 符合性声明;
● 医疗器械描述;
● 设计验证与确认文件总结:
○ 临床前研究总结(包括灭菌验证和货架寿命研究,如适用);
● 临床评估报告总结;
● 拟用医疗器械标签;
● 风险分析;
● 制造商信息:
○ 制造场所和灭菌场所的名称与地址;
○ 至少符合下列质量管理体系之一的证明:
⊕ ISO 13485;
⊕ MDSAP;
⊕ 符合美国FDA质量体系法规;
⊕ 日本厚生劳动省第169号法令;
● 制造工艺流程图。
补充文件
● 授权书;
● 医疗器械配置清单;
● 获得一个独立的海外参考监管机构批准的证明(例如,批准信函、证书);
● 对于含有药品成分的医疗器械,如需符合简化注册程序,则适用以下条件:
○ 作为一个医疗器械已获得一个独立的海外参考监管机构批准的证明;
○ 证明其化学或生物成分已获得至少一个世界卫生组织认可的主管药品监管机构评估和批准的证明。
4.B、C、D类医疗器械如何注册
您需要通过新加坡卫生科学局医疗产品注册与管理电子系统(SHARE)进行医疗器械注册。您需要具备以下条件才能访问SHARE系统:
● 企业通行证(CorpPass)。
对于优先审评计划的申请,在SHARE平台提交产品注册申请时,选择“优先审评计划”。
如果您需要在新加坡进行医疗器械注册或医疗相关牌照的申请,欢迎扫描下方二维码咨询金阁顿专业顾问。金阁顿官方合作伙伴(包括合资公司)在新加坡、印度尼西亚及马来西亚依法持有相关医疗牌照与资质,可协助医疗器械及医药品牌出海,以推进国际市场布局。
此外,金阁顿可为您提供一站式专业服务,全面支持医疗器械与医药品牌出海的全流程需求。

金阁顿(GolddenGroup)成立于花园城市新加坡,专业服务高净值家族和专业金融机构。主打业务有新加坡家族办公室/家族基金设立,私募基金(VCC及子基金)备案,PPLI,境外资产管理,新加坡移民,和境外保单,是您新加坡一站式的家族管家。金阁顿,为您的下一代继续服务。

·【私募人寿保险(PPLI)】(下)从中国大陆到新加坡:PPLI如何补足离岸公司与
·【私募人寿保险(PPLI)】(中)透明时代下的财富架构重构:PPLI如何补足信托
·【私募人寿保险(PPLI)】(上)税收透明时代的财富持有:PPLI如何实现递延增


 info@golddengroup.com
info@golddengroup.com  152 Beach Road,#20-01,Gateway East,189721
152 Beach Road,#20-01,Gateway East,189721 